


Since the implementation of the CARES Act reporting requirement, the question continues to be asked: Is it really a requirement? The answer is yes, and now it is no longer recommended, but is stated as “should” in the newly published Final version of the Guidance for Industry (GFI) “Reporting Amount of Listed Drugs and Biological Products Under Section 510(j)(3) of the FD&C Act” (February 2024) (here).
On February 5, 2024, the FDA finalized the GFI on what has become known as “CARES Act reporting” or Section 510(j)(3) reporting. Because of the late publishing of this Final GFI, the reporting for Calendar Year (CY) 2023 should be submitted No Later Than August 5, 2024. Reporting in subsequent years (starting in CY 2024) should be submitted no later than March 31 of the following year. Additionally, if you have not submitted your reports for CY 2020, 2021, and/or 2022, they should be submitted to the Agency as soon as possible.
In addition to the change in reporting timing, there are several other key changes from the October 2021 Draft GFI of the same name. We suggest you thoroughly review the entire GFI but we have highlighted what we think are the most significant changes here:
- The FDA expanded the definition of a “listed drug” that must be reported to include drug product that is not in finished package form. It further discusses how this is to be reported in Section III.B.5.a.ii., along with examples in Table 3. Additionally, it is covered in the response to Section IV. Questions and Answers, Question A (Q&A IV.A).
- In Footnote 6, the FDA added the statement that “Other listed unapproved drugs are also subject to reporting under Section 510(j)(3) of the FD&C Act” when speaking of certain nonprescription drug products that may be lawfully marketed without an approved application, such as Over-the-Counter monograph drugs and animal drug products that are not approved, conditionally approved, or indexed. Section III.B.5.b. details how this is to be done.
- The FDA clarified that the obligation to report pertains to registrants and not the application holder unless they are also the registrant. However, it also states that applicants may do the reporting for the CMO as an authorized agent. (See also, Q&A IV.D.)
- In Footnote 12, the Agency clarified that donated drugs and drug samples are considered to be in commercial distribution.
- The FDA expanded on the background to explain how it will use the data and why the Agency is requesting it from multiple places in the supply chain. The FDA is using this to be better informed so that it can be better able to promote stronger supply chains and reduce drug shortages, but also to inform the Agency regarding the need for surveillance inspections, based on potential public exposure to a drug manufactured at a given facility. (See also, Q&A IV.C.)
- Additionally, the Agency confirmed that the data reported is generally expected to fall in the category of confidential commercial information (CCI) and, therefore, exempt from disclosure to the public.
- Section III added sections on Who Must Report and What to Report (which contain some information from the previous “Content of Reports”):
- The FDA now directs registrants to consider when their drugs were manufactured and suggest this is defined as the month in which the drug is released.
- The FDA also provide further definition on what it means when it states that the product is manufactured for commercial distribution. Specifically, the Agency states that this should include:
- For domestic establishments, drug manufactured for commercial distribution either within, or outside of, the U.S., as long as it is not for investigational use only, or an interplant transfer between registered establishments under common ownership and control.
- For foreign establishments, commercial distribution has the same meaning except that it does not include distribution of product that is not imported nor offered for import into the U.S. The Agency further clarifies that, if a drug is manufactured that may be offered for distribution into the U.S., but also complies to multiple countries’ standards, even if it is not imported into the U.S., it should be reported. (see also, Q&A IV.I)
- Lastly, the FDA clarifies that if an establishment manufactures both the API and the finished dosage, and the API is consumed at the establishment for the finished dosage manufacture and not for sale or commercial distribution, then it is not required to be listed, and therefore does not have to be reported under Section (j)(3).
- Regardless of whether multiple manufacturing activities are performed at a given establishment, the business operation should match the single business operation included in the drug listing for that establishment. The Agency further differentiates that a contract manufacturer should provide the amount of drug that they manufacture themselves, not taking into account further processing that may occur. (See also, Q&A IV.K.)
- For human drug products, Section 510(J)(3) reporting must be done for the NDC associated with the registrant’s labeler code as well as for the NDC associated with a Private Label Distributor (PLD). Each is to be reported separately under each NDC (each labeler code).
- However, for animal drugs manufactured for commercial distribution under a trade name or label of a private label distributor, the drug must be listed only under an NDC associated with the PLD’s labeler code.
The FDA has added the following diagram to help determine what and how to report:
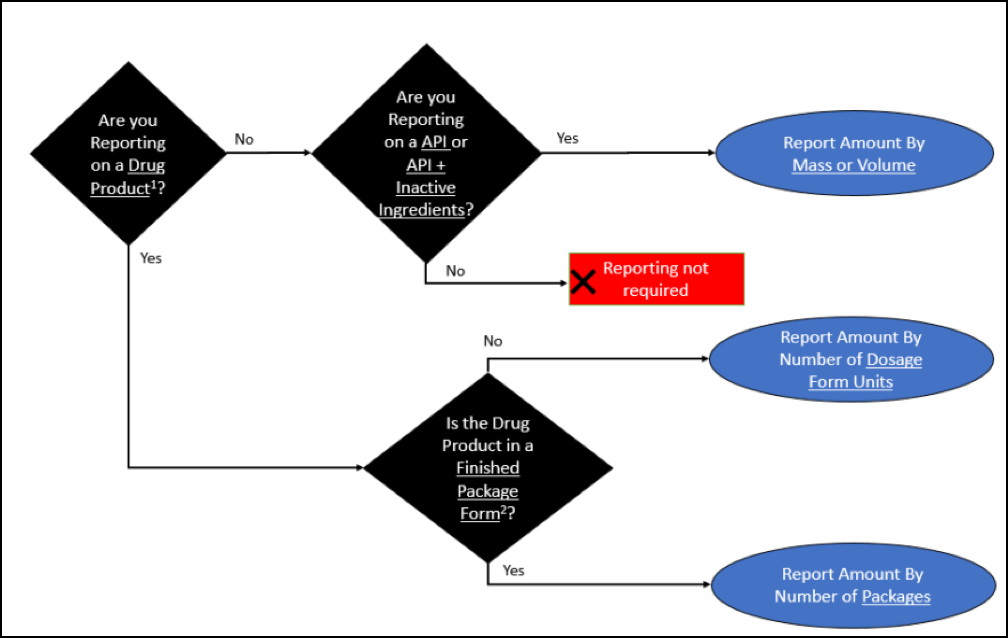
1 For the purposes of this guidance, drug product means a finished dosage form, for example, tablet, capsule, solution, etc., that contains an active drug ingredient generally, but not necessarily, in association with inactive ingredients (see 21 CFR 210.3(b)(4)).
2 For the purposes of this guidance, finished package form means a form suitable for distribution to pharmacies, hospitals, or other dispensers or sellers of the drug product to patients or consumers.
(from Final GFI “Reporting Amount of Listed Drugs and Biological Products Under Section 510(j)(3) of the FD&C Act” (February 2024).)
The Q&A section referenced above contains the same questions as in the Draft GFI; however, some of the responses are amended with additional information.
Hopefully the Final GFI will further clarify any remaining questions that your company had regarding reporting. However, if you have any questions regarding CARES Act reporting or need assistance doing so, please contact r.welton@lachmanconsultants.com. Happy Reporting!
