
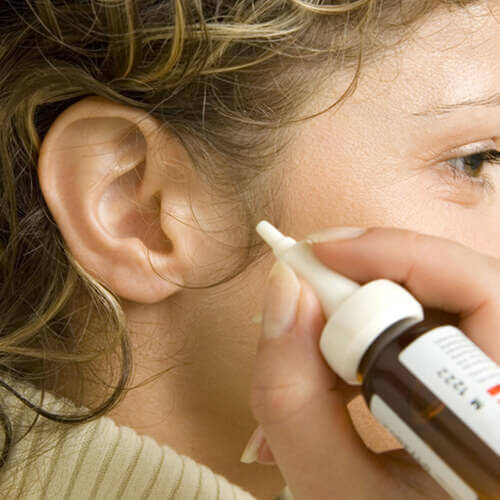
For years, FDA has been indicating that it was planning to take enforcement action against certain marketed unapproved prescription and OTC ear drops, but, yesterday, the final shoe dropped on the marketing of such products, as FDA published its intent to take enforcement action against these products in a Federal Register notice (here).
The FDA discussed certain adverse events, some of which were serious, that have been reported to be associated with these products (especially in pediatric patients) and have concluded that the continued marketing of these products pose a risk to public health. In addition, FDA notes that the continued marketing of these unapproved products pose an affront to the drug review and approval process and/or are in conflict with the existing OTC monograph.
The Agency further states that
“FDA has reviewed the publicly available scientific literature for the following unapproved prescription drug products: (1) Single-ingredient otic drug products containing benzocaine; (2) fixed-dose combination otic drug products containing benzocaine and antipyrine; (3) fixed-dose combination otic drug products containing benzocaine, antipyrine, and zinc acetate; (4) fixed-dose combination otic drug products containing benzocaine, chloroxylenol, and hydrocortisone; (5) fixed-dose combination otic drug products containing chloroxylenol and pramoxine; and (6) fixed-dose combination otic drug products containing chloroxylenol, pramoxine, and hydrocortisone. In no case did FDA find literature sufficient to support a determination that any of these prescription products are generally recognized as safe and effective. Therefore, these prescription drug products are “new drugs” within the meaning of section 201(p) of the FD&C Act (21 U.S.C. 321(p)), and they require approved new drug applications (NDAs) or abbreviated new drug applications (ANDAs) to be legally marketed.”
As far as any discretion relative to enforcement action, the FDA outlines its intent in the notice (see below), but also notes that they do not plan to issue separate Warning Letters before taking enforcement action.
“Any drug product covered by this notice that a company (including a manufacturer or distributor) began marketing after September 19, 2011, is subject to immediate enforcement action. For products covered by this notice that a company (including a manufacturer or distributor) began marketing on or before September 19, 2011, FDA intends to take enforcement action against any such product that is not listed with the Agency in full compliance with section 510 of the FD&C Act (21 U.S.C. 360) before July 1, 2015, and is manufactured, shipped, or otherwise introduced or delivered for introduction into interstate commerce by any person on or after July 1, 2015. FDA also intends to take enforcement action against any drug product covered by this notice that is listed with FDA in full compliance with section 510 of the FD&C Act but is not being commercially used or sold6 in the United States before July 1, 2015, and that is manufactured, shipped, or otherwise introduced or delivered for introduction into interstate commerce by any person on or after [INSERT DATE OF PUBLICATION IN THE FEDERAL REGISTER]. However, for drug products covered by this notice that a company (including a manufacturer or distributor): (1) Began marketing in the United States on or before September 19, 2011; (2) are listed with FDA in full compliance with section 510 of the FD&C Act before July 1, 2015 (“currently marketed and listed”); and (3) are manufactured, shipped, or otherwise introduced or delivered for introduction into interstate commerce by any person on or after [INSERT DATE OF PUBLICATION IN THE FEDERAL REGISTER], the Agency intends to exercise its enforcement discretion as follows: FDA intends to initiate enforcement action regarding any such currently marketed and listed product that is manufactured on or after [INSERT DATE 45 DAYS AFTER DATE OF PUBLICATION IN THE FEDERAL REGISTER], or that is shipped on or after [INSERT DATE 90 DAYS AFTER DATE OF PUBLICATION IN THE FEDERAL REGISTER]. Furthermore, FDA intends to take enforcement action against any person who manufactures or ships such products after these dates. The purpose of these enforcement timeframes is to allow manufacturers and distributors to deplete their current inventory and ensure a smooth transition for consumers. Any person who has submitted or submits an application for a drug product covered by this notice but has not received approval must comply with this notice.
The Agency, however, does not intend to exercise its enforcement discretion as outlined previously if either of the following applies: (1) A manufacturer or distributor of drug products covered by this notice is violating other provisions of the FD&C Act, including, but not limited to, violations related to FDA’s current good manufacturing practices, adverse drug event reporting, labeling, or misbranding requirements other than those identified in this notice or (2) it appears that a firm, in response to this notice, increases its manufacture or interstate shipment of drug products covered by this notice above its usual volume during these periods.”
