

On June 11, 2019, the FDA held a CDRH webinar on the Q-Submission Program for Medical Device Submissions. The webinar was based on the recent May 7, 2019 Final Guidance document titled “Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program.” This guidance document supersedes “Requests for Feedback on Medical Device Submissions: Pre-Submission Program and Meetings with FDA and Staff.”
So, what is a Q-Submission? The Q-Submission process (Q-Sub for short) is a path to request different types of medical device submission-related communications with the FDA. This new guidance is a refined process on how communications can be set up with the FDA, in comparison to the previous documents. In 1995, there was the Pre‑IDE feedback system, then, in 2013, the FDA launched the Pre-Submission Program and expanded Pre‑IDE communications to include other marketing submissions, such as Pre‑PMAs and Pre‑510Ks. Now, in 2019, the Q-Sub process has widened its reach to include a broad scope of other medical device-related interactions (including informal interactions). The following chart was taken from one of the slides (presented by Susannah Gilbert, Acting Policy Analyst, CDRH) and summarizes the types of Q-Subs that can be requested:
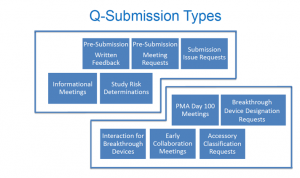
Important statements were made by FDA during the webinar regarding:
- Pre-Submission Communications: The FDA stressed the importance of preparing specific questions (precision vs. ambiguity) in advance and recommend three to four topics as most. Verbal meetings are expected to last no more than an hour. The company is required to take minutes and the FDA promises to have feedback on the minutes within thirty days.
- Submission Issue Requests (SIRs): You can request written feedback or a meeting (but not both!). SIRs are intended to facilitate interaction between the FDA and the submitter to quickly resolve (or clarify) issues identified in these letters so that projects can move forward. Submitters are expected to provide a formal response to the FDA within the required timeline.
- Study Risk Determinations: The FDA will provide a final decision in writing to determine the one of four levels of risk for the proposed clinical study.
- Informal Meetings: These are opportunities to have dialog with the FDA on topics such as device development, new technologies (e.g., artificial intelligence and SaMD were discussed in the question and answer period), and other topics outside the scope of the Q-Subs mentioned. Informational Meetings are requests to share information with the FDA without the expectation of official feedback (which is described in the guidance document as “information sharing”).
- The types in the lower portion of the chart above: The Q-Sub program provides a mechanism to track interactions described in other FDA program guidance documents.
There is one thing that was not mentioned during the conference call, but is key information that combination product companies should know: The guidance document states that questions for these products may take longer for response (up to seventy-five days) due to factors such as the increased number of Agency staff involved and other associated regulatory complexities. This is in alignment with my own experience in the combination products industry, and I advise that you engage the FDA very early in the development process if you have any regulatory questions (timing is key!).
The rest of the webinar spent time discussing significant changes from the previous guidance, with newly established timeframes for MDUFA IV commitments for pre-submissions and updated timing on SIRs.
In summary, I recommend you review the slides of the webinar and the guidance document located on the FDA website at the links below. If you are a new product developer, the earlier you submit for FDA communications, the better it will be for your organization to get clarity and be well on your way to getting your products on the market!
- Printable Slides from the June 11, 2019 Webinar (presented by Susannah Gilbert, Acting Policy Analyst, Division of Submission Support Office of Regulatory Programs, CDRH)
- Q-Sub Guidance Document (May 7, 2019 Final FDA Guidance “Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program”)
