

Irritation has been building between the Office of Generic Drugs (OGD) and industry over the use of the terms backlog vs. workload, particularly over the last few months. At the recent Annual Meeting of the Association for Accessible Medicines (AAM), formerly the Generic Pharmaceutical Association (GPhA), the semantics pot almost boiled over. So why the big debate over the term describing the number of ANDAs OGD has in its court, whether they are original un-reviewed applications or those where a Complete Response Letter has been answered by the applicant, or those that are awaiting response by industry (more on this later)? Well, some of the argument relates to history and some of it (I assume) relates to optics.
Let’s start with the historical view. When Hatch-Waxman first passed in September of 1984, there was a brief 60-day period before the statute took effect. On November 24, 1984, the first day that the then Division of Generic Drugs (note – for clarity’s sake we will call it OGD throughout the rest of this post) could receive an ANDA, trucks literally lined up at the loading bay of the Parklawn building in Rockville, Maryland and well over 1,000 ANDAs were shuffled upstairs to a small staff of dedicated federal employees just waiting to see what was to befall them. Having been there at the time, I can tell you it was nothing like we expected. From that date forward, we had 180 days to review each application and we called it our backlog. Why? Not because the applications were overdue (although after the first 180-days past some were), but because it was work that we had not yet completed. The term backlog was applied consistently over the years to mean just that – an ANDA in some stage of OGD review where the review had not yet been completed and a review letter, approval, or tentative approval letter had not yet been sent to the sponsor.
Even a review of the last “old format” Monthly Statistical Report of August 2013 (below) prepared by “OGD” (which was the last such formatted report before GDUFA I changed the FDA’s and industry’s world), we can see it reflected the number of “Original ANDAs Pending Per Month” and included new ANDAs and those where a firm had responded to what were then called deficiency letters.
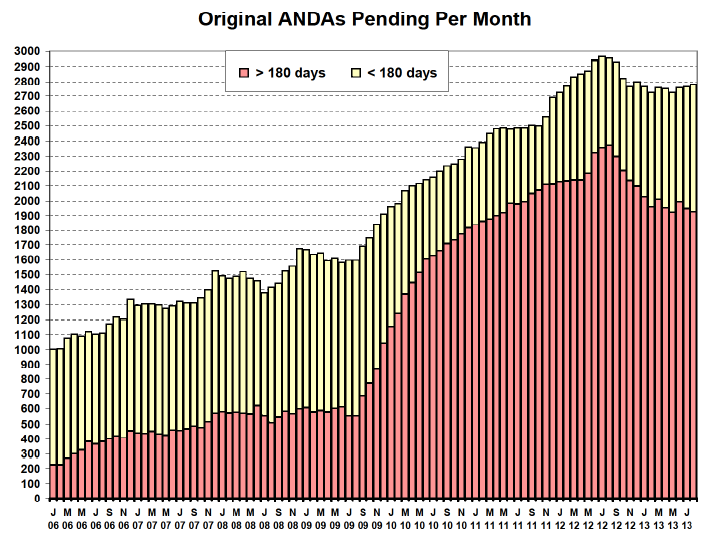
As you can see, those in red were over the statutory 180-day review time period and those in yellow had not yet hit the 180-day review clock. However, they were all lumped together for reporting what we then called the “backlog”. Progress was measured by the reduction in the number of ANDAs over the 180-day review period and, generally, by a decrease in the total number of ANDAs pending in any given month.
A dramatic shift came with the passage of GDUFA as OGD began a transformational change in the ANDA review and approval process. However, in addition to not only process changes, GDUFA provided a statutory definition to the term “backlog applications” and assessed a separate fee called a “backlog fee” to those applications pending at OGD or back with industry for a response to a deficiency letter prior to October 1, 2012, the implementation date for GDUFA. It was then that things began getting a little bit fuzzy whenever the FDA and industry talked about the “backlog”. Some continued to use the term in its statutory sense under GDUFA – or as stated in the GDUFA Commitment letter as those applications pending as of October 1, 2012 – while industry observers used the same term to describe all ANDAs pending final Agency action, once their cohort year metric date passed. This caused considerable confusion despite the pains some Agency officials took to clarify which meaning was intended. Now with the rapid approach of GUDFA II, there appears to be more of an issue of optics and perception driving the tension between the use and context of the terms “backlog” and “workload”.
As we know, FDA has met or exceeded all its GDUFA I goals to date. But still when anyone outside of FDA writes or discusses submitted, but unapproved ANDA applications, they call it the backlog. OGD argues that FDA’s medical product user fee programs distinguish between submissions awaiting FDA action and submissions awaiting applicant action. From FDA’s perspective, substantially all ANDAs at FDA are within the GDUFA review goal or Target Action Date (TAD) and most of them have been reviewed at least once. Moreover, a significant number of ANDAs are with applicants and not the Agency. As noted in a previous blog post, many of the ANDAs pending with applicants are likely to come back into the FDA’s court as delayed, due to prioritization of the ANDAs by the applicants. In some instances, the ANDAs may no longer be viable to pursue, but they are often counted as part of the backlog and FDA has little control or insight as to their true impact on workload. But again, isn’t this mostly semantics? FDA and industry share the goal of approving more generics faster and getting to a “steady state” where output matches input.
Drug pricing issues are being hotly debated in Congress and are front page news in the press. Clearly, one way to lower drug prices is to approve more generic drugs. So, whenever a generic version of a product is approved it should, and likely does in some manner, contribute to either stabilizing or lowering drug prices for that specific product. Does it matter if the approval comes from something called the “workload” or the “backlog”? No, it does not. This is where the discussion turns into one of optics– and in Washington, everything has to do with optics. But should we be expending our energy in defining the ANDAs pending before OGD or should we just decide to define a single term that everyone understands?
Perhaps we can look at it this way. One might think of the pre-GDUFA ANDAs as cohort year 0, and the GDUFA cohort years ANDAs as falling into cohort years 1-5. If we see statistics that make actions on these 6 cohorts transparent, and we can understand where the resources are being expended and the progress of each cohort year’s applications over time, why should we care if those applications fall into what is called the “backlog” or “workload” applications? The answer is- we should not!
So where do we go from here? We need to get past using any particular term and instead focus the discussion toward ongoing progress and results for each of the cohort years 0-5. The ultimate goal is not to have zero pending applications – that would mean that industry is not submitting new applications, and this would serve nobody’s interests but the innovators. In order to increase the supply of affordable generic medicines, the generic industry absolutely must continue submitting new applications. Instead, the ultimate goal should be to have a transparent, predictable review process where high-quality applications are reliably reviewed – and, hopefully, approved – within a 10-month timeframe.
It is time that we concentrate on moving the industry and FDA towards this desirable steady state. This will require developing a common understanding of what is reasonable from both industries and the Agency’s perspective, taking into account the entire end-to-end process. The process begins during a generic company’s R&D phase, runs through the development and submission of a high-quality application, includes the Agency’s review of that application, and eventually yields an approval of a safe and effective drug product that can reach the market in the timeliest manner. This will require the Agency to provide clear standards for those areas where industry needs timely and consistent guidances; it will require that industry take great pains to provide the information the Agency needs to conduct its review of an application; and it will require that both sides stay focused on collaboratively working out any scientific, regulatory, or manufacturing issues. We need simplicity and transparency. We need faith in the process and we need to adequately resource the Agency, as well as be certain that industry is providing the highest quality applications to OGD for review. There needs to be movement on both sides to accomplish this goal and the parameters need to be clear and definitive. The good news is, that in her remarks at the AAM Annual Meeting, Dr. Uhl signaled enthusiasm for working with industry on these issues and stressed that FDA and industry are aligned on the objective of reaching steady state.
Towards that end, AAM and its member companies insisted on robust, detailed performance reporting as part of the proposed GDUFA II agreement. For example, monthly, FDA must report inputs (submissions) versus outputs (approvals and other actions). Quarterly, FDA must report the number of ANDAs awaiting FDA action and the number of ANDAs awaiting applicant action. Annually, FDA must report mean and median approval times and mean and median number of ANDA review cycles to approval by Fiscal Year receipt cohort. Rest assured that I will watch the data like a hawk, and report on FDA’s progress towards the desired steady state, regularly.
