

As a group of consultants to domestic and international pharmaceutical companies, we work with many clients responding to inspectional findings from an FDA inspection. At the close of the inspection, an FDA-483, Inspectional Observations form, may be issued to the firm’s management outlining conditions, which the FDA investigator(s) believe may constitute deviations from applicable law or regulations. FDA investigators provide guidance to industry stating that a response to the 483 is encouraged within 15 days after the close of the inspection, if the firm would like the response considered prior to the Agency making any decision on resulting action, specifically, the issuance of a Warning Letter.
The FDA’s Regulatory Procedures Manual states that the District office should make a recommendation to the appropriate reviewing office within 15 days of the close of the inspection or conclusion of sample analysis. Within 15 days, the reviewing office should notify the District of its decision regarding the recommendation.
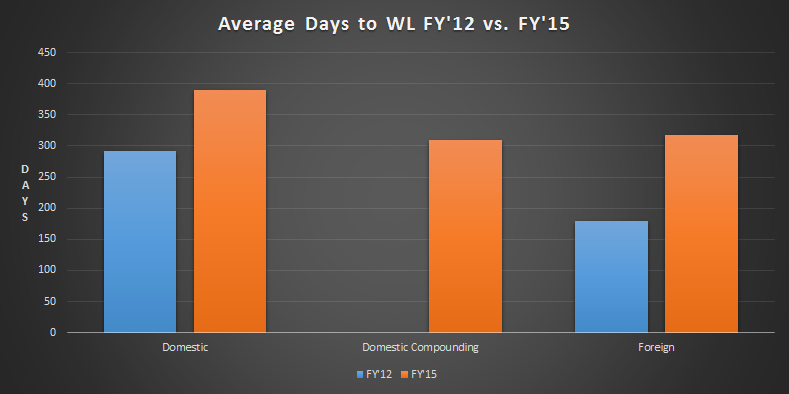
In 2012, the average time for a Warning Letter to be issued was 292 days for a domestic inspection and 179 days for a foreign inspection. By end of fiscal year 2015, the average time for issuance was 390 days for a domestic, non-compounding facility and 310 days for a domestic, compounding facility. The time for foreign inspection issuance had increased to 317 days.
The delay in receiving formal notice and feedback from the Agency regarding Inspectional Observations, makes it challenging for industry to understand the level of FDA concern regarding their operations and the Agency’s determination of the relevancy of the investigator’s findings. While industry receives the FDA-483 at the close of the inspection, the supporting detail, relevance, and evidence is not available to industry until the copy of the investigator’s establishment inspection report (EIR) is released. It is expected that industry will immediately address observations presented by the investigator, often during the course of inspection. The Warning Letter provides direct reference to the section of the Act and the specific regulation for which the firm did not comply, allowing their quality team and/or consultants to ensure they are pursuing proper and expected corrective action.
So why is it taking months, and sometimes years, for the Center for Drug Evaluation and Research (CDER) in conjunction with the District office to issue a Warning Letter? One possible explanation is the Agency’s staffing shortage. This shortage is not only in the District, but also in CDER. FDA is actively recruiting investigative staff to help perform this work, as can be seen through national job announcements. Further, FDA’s 2017 budget includes 3,800 additional staff across the Centers alone.
The June 2016 ISPE Quality Manufacturing Conference in Bethesda, MD brought opportunity for current and ex-FDA staff to discuss the matter through educational sessions and networking. FDA recognizes the issue and states that they are working on a resolution to provide more timely responses.
Presentations provided by current FDA Office of Regulatory Affairs (ORA) senior leaders at the 120th Association of Food and Drug Officials Annual Educational Conference in Pittsburgh, PA, clarified that that the Agency is dedicated to hiring additional staff and continuing improvements in collaboration between the Centers and ORA. We are sure that the pharmaceutical industry worldwide would welcome continued feedback and opportunity for dialogue with FDA on this matter and the impact of the delay on industry compliance.
